Description:
The
program receives as input a pair of PDB files (or uploaded files in PDB
format) and finds "large enough" geometrically similar substructures in
both molecules, which can be rigidly superimposed. It returns a set
of hypotheses sorted by the size of the superimposed substructures.
The method superimposes the C-alpha atoms of the proteins and is sequence-order
independent.
Program url:
Examples: Bacterial electron transferring proteins
1. (Almost identical structures) 1pmy with 1pza
Sequence Alignment:
Score
= 116 bits (274), Expect = 8e-26 Identities = 54/120 (45%), Positives
= 75/120 (62%)


2. (more distant in sequence) 1pmy with 1aaj
Sequence Alignment:
Score
= 32.1 bits (71), Expect = 2e-06 Identities = 20/78 (25%), Positives
= 42/78 (53%), Gaps = 3/78 (3%)
Structural matching:
Result 1: Match size: 78 RMS: 1.44
Rotation: 1.178 -0.059 -2.615 Translation: 30.230 14.864 17.912
Rotations are given in radians for X-Y-Z axes. Rotating space around the X-axis, then around the Y-axis and finally
around
the Z-axis would give the required rotation. X-Y-Z translation coordinates
are given in Angstrom units.


Note the similar and the different parts of the two proteins.
Note
insertions/deletions in loops.
3. (same fold, distant structures) 1svy with 2vik
Both are gelsolin (actin depolymerizing) proteins from slime mold and chicken, respectively.
Sequence Alignment:
(Note,
that without the structural information, sequence based alignment can be
different than structural alignment!)
Score = 18.8 bits
(37), Expect = 0.025
Identities = 23/67
(34%), Positives = 30/67 (44%), Gaps = 12/67 (17%)
Query: 29 VPVPTLSYGNFYEGDCYVLLSTRLTGSGFSYNIHYWLGLNSSQDEQGAAAIYTTQMD-EY
87
VP+ T S + GDC+ LL LT
I+ + G SS E AA +D E
Sbjct: 20 VPLATSSLNS---GDCF-LLDAGLT-------IYQFNGSLSSPQELNLAAEVARAIDAER
68
Query: 88 LGSVAVQ 94
LG V+
Sbjct: 69 LGLPLVE 75
Structural Alignment:


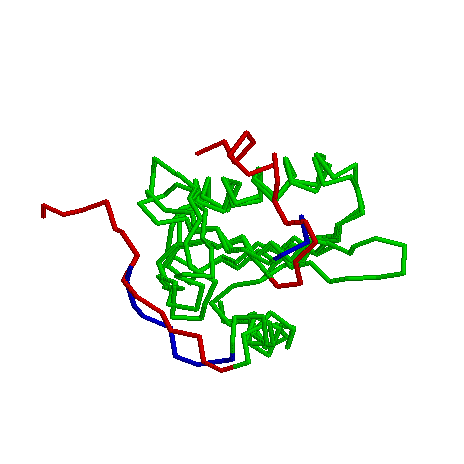
To watch only the structurally aligned part, look at 2vik:18-111, 1svy:250-161 (green color).