Multiple sequence alignment
 Introduction
Introduction
FASTA
BLAST
FLASH
 Multiple sequence alignment
Multiple sequence alignment
So far we have only considered methods to align two sequences. However,
when the sequence data is available, a multiple alignment is always preferable
to pairwise alignment.
There are number of techniques for the alignment of three or more sequences
calculations.
Dynamic programming Hyperlattice
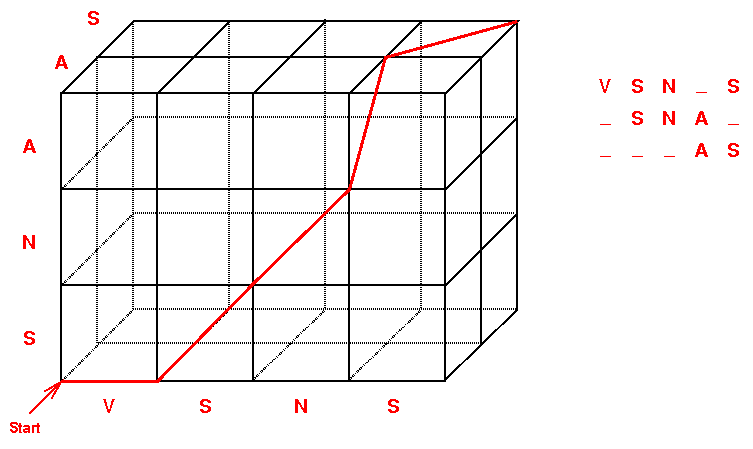
We can denote a path through a lattice in a simple way. For each node
visited, we list, component by component, the distance from the starting
point in the bottom left (i.e. from the source (0,0,0) of the lattice).
For example, the path in Fig. 1 is written down as (0,0,0), (1,0,0), (2,1,0),
(3,2,0), (3,3,1), (4,3,2). As you can see, the distance from the starting
point is the number of letters already aligned. For example, column 4 of
the alignment corresponds to node (3,3,1). Indeed, 3 letters from the first
and second sequence are aligned at that point, and one letter from the
third.
Hierarchical extensions of pairwise methods
Practical methods for multiple sequence alignment based on a tree. The
principle is that since the alignment of two sequences can be achieved
very easily, multiple alignments should be built by the successive application
of pairwise methods.
This is by far the most practical and widely used method.
The steps are summarized here :
- Compare all sequences pairwise. For N sequences
there are [Nx(N-1)]/2 pairs.
- Perform cluster analysis on the pairwise data to generate a hierarchy
for alignment. This may be in the form of a binary tree, or a simple ordering.
- Build the multiple alignment by first aligning the most similar pair
of sequences, then the next most similar pair and so on. Once an alignment
of two sequences has been made, then this is fixed. Thus for a set of sequences
A, B, C, D having aligned A with C and B with D the alignment of A, B,
C, D is obtained by comparing the alignments of A and C with that of B
and D using averaged scores at each aligned position.

This family of methods gives good usable alignments with gaps it can
be applied to large numbers of sequences, and with the exception of the
initial pairwise comparison step is very fast.
Although based on the successive application of pairwise methods, multiple
alignment will often yield better alignments than any pair of sequences
taken in isolation.
Back to index
FASTA
Modern databases contain tens of thousands sequences, among them there are
very long sequences (tens and hundreds of thousands symbols). It makes
a dynamic programming database scan rather difficult. Accordingly, Pearson
and Lipman developed the program FASTA for sequence scans that in concept
searches for the most significant diagonals. The initial step in the algorithm
is to identify all exact matches of length k (k-tuples) or
greater between two sequences. For example, for proteins, if k =
3 then there are 8000 (203) possible k-tuples and each
element of an array C of length 8000 is set to represent one of
these k-tuples. Sequence A is scanned once and the location
of each k-tuple in A is recorded in the corresponding element
of C. Sequence B is then scanned and by reference to C
the location of all k-tuple matches common to A and B
may be identified. FASTA saves the 10 highest regions of identity which
are then re-scored with PAM250 matrix. If there are several initial regions
above a preset cutoff score then those that could form a longer alignment
are joined, allowing for gaps and a score initn is calculated by
subtracting a penalty for each gap. initn is used to rank the database
sequences by similarity. Finally, dynamic programming is used over a narrow
region of the high scoring diagonal to produce an alignment with score opt.
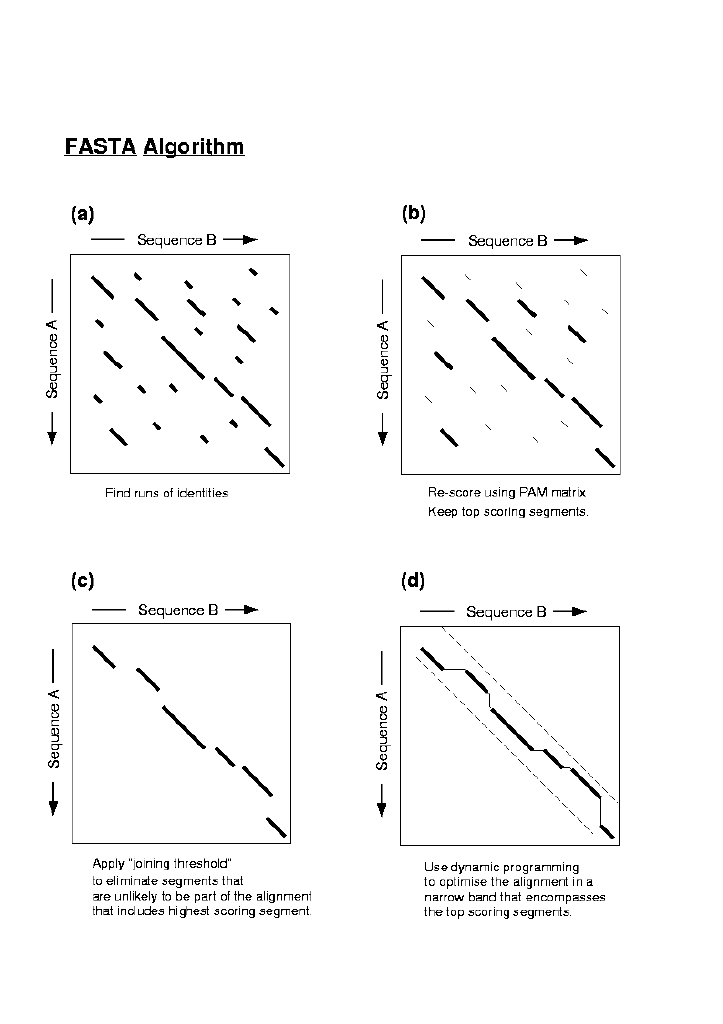
FASTA only shows the top scoring region, it does not locate all high
scoring alignments between two sequences.
Back to index
BLAST
- Basic Local Alignment Search Tool
BLAST is heuristic method to find the highest locally optimal alignments
between a query sequence and a database. The important simplification that
BLAST makes is not to allow gaps, but the algorithm does allow multiple
hits to the statistics of the sequence alignments without gaps by Karlin and Altschul.
The statistics allows the probability of obtaining an alignment without gaps
(HSP - Highest Segment Pair) with a particular score to be estimated. The
BLAST algorithm permits nearly all HSP's above a cutoff to be located efficiently
in a database.
The algorithm operates in three steps:
1. For a given word length k (usually 3 for proteins) and score
matrix a list of all words (k-mers) that can score > T
when compared to k-mers from the query is created.
2. The database is searched using the list of k-mers to find
the corresponding k-mers in the database (hits).
3. Each hit is extended to determine if an HSP that includes the k-mer
scores > S, the preset threshold score for an HSP. Since pair
score matrices typically include negative values, extension of the initial
k-mer hit may increase or decrease the score. Accordingly, a parameter
X defines how great an extension will be tried in an attempt to
raise the score above S. 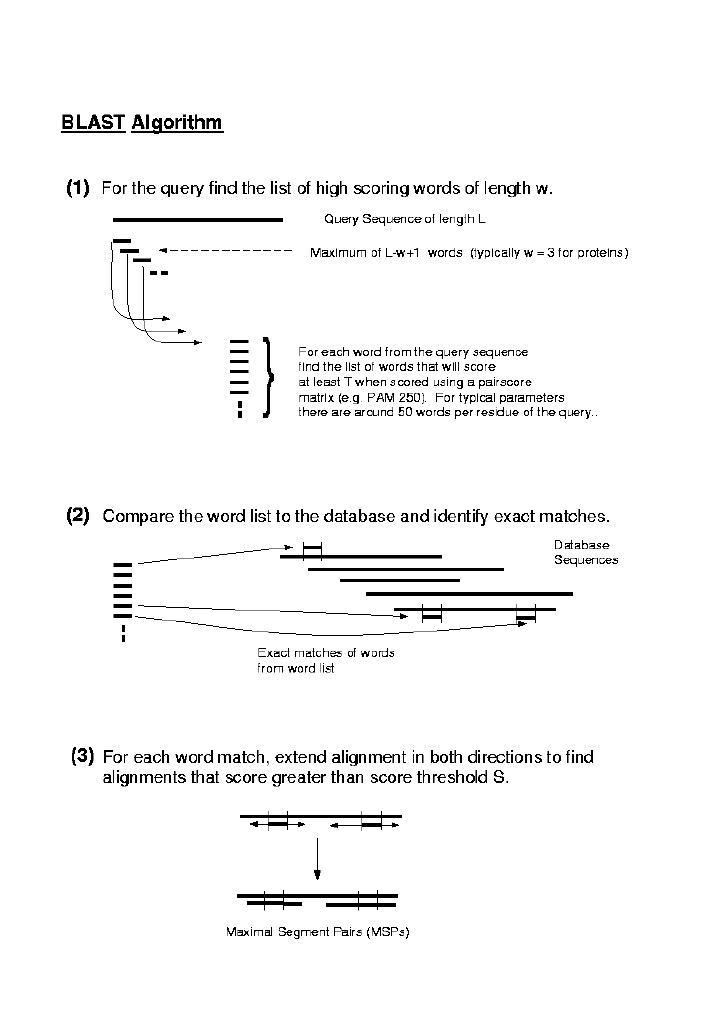
A low value for T reduces the possibility of missing HSPs with
the required S score, however lower T values also increase
the size of the hit list generated in step 2 and hence the execution time
and memory required.
BLAST is unlikely to be as sensitive for all protein searches as a full
dynamic programming algorithm. However, the underlying statistics provide
a direct estimate of the significance of any match found.
Back to index
FLASH
- A Fast Look-up Algorithm for String Homology
Recently, Rigoutsos and Califano of the IBM T.J. Watson Research Center
have extended the idea of indexing to allow for gaps and mismatches (substitutions).
The indexes, or lookup tables are highly redundant and based on a probabilistic
model. As a consequence the index files are very large and the problem
is less one of absolute CPU speed, and more a question of fast disk access.
However, the Rigoutsos and Califano FLASH algorithm permits very rapid
scans to be performed on protein databases with claimed sensitivity and
accuracy close to dynamic programming.
Back to index