Introduction to Protein Comparison
3-D Protein Matching
Problem
Problem definition
Applications
Examples
Geometric Hashing
Flexible Matching
Problem:
Detection of a-priori unknown substructure which are almost isometric
modulo the 3-D rigid motion group.< >
Problem definition:
Given A known Database of molecules, each as
a set ( linearly ordered, or not) of points representing Atoms ,
and given a new Molecule M: Find all the molecules Mi
in the DB, and rigid Transformations Ti : R3
-> R3 so that a matching from ( a part of ) the Atoms of M
to those of Mi will give an RMSD (root mean square distance) bellow a given
limit L.
Applications:
-
structural comparison of proteins
-
receptor ligand docking
Back to Index.
Examples
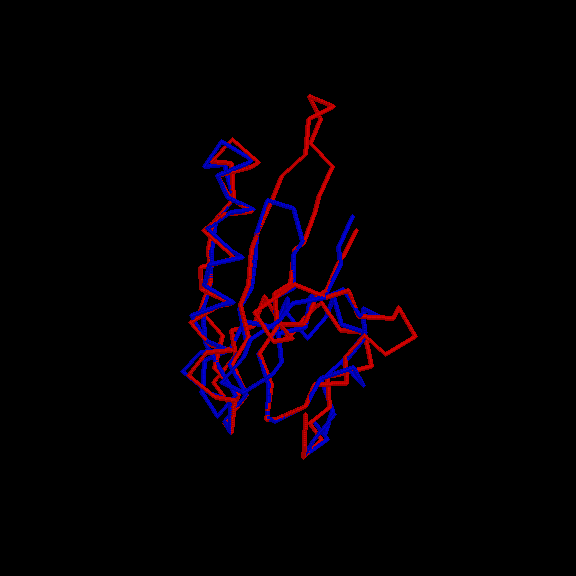
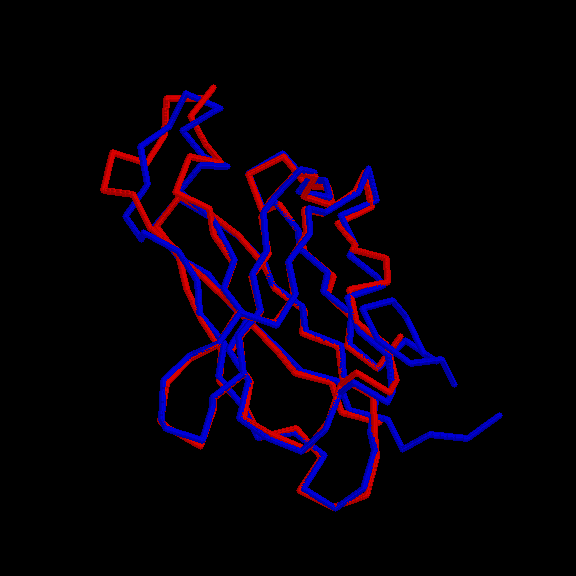
Back to Index.
Geometric Hashing:
the 2D case:
For each ordered pair of points (a,b) in the plane there exists an unique
Coordinate system (Frame of Reference) such that its center is on the first
and its x axis is the vector from b to a. If we take two such points in
a model, and calculate the coordinates of all the other points in this
reference frame, then , these coordinates will be invariant under any translation,
rotation ,similarity, or a combination of these three kinds of transformations.
More over, these coordinates will only dependent on choosing the right
base pair ,i.e. if we show only some of the points of the model (partial
overlapping) then, given the right base pair, we will still identify the
remaining points. It seems logical, then, to store these frames of reference,
and coordinates within them, in some Fast Retrieval Data Structure, so
that given a new scene, they will be used immediately, so that we will
concentrate only on transformations likely to occur.
This gives us the GH Algorithm for the 2D case:
we will divide the work into two main stages:
-
Preprocessing stage:
For each model (2D "molecule") we iterate on all pairs of its points
("Atoms") .
For each basis pair we:
Compute the associated frame of reference.
For each point in each model we:
Compute its coordinates in the above frame of reference Use the coordinates
as a key for inserting an Entry to a Hash table.
The Entry will include:
{
Model name.
Frame of Reference ( the basis pair, not the f.o.r. itself)
Identity of point in model
}
The complexity of this stage is O( N3 * M) where M is the number
of models and N the number of points in each model.
This stage requires no knowledge of the "Scene" (the input "molecule")
and can be done off line.
-
Recognition stage:
Given a scene:
For each ordered pair of points:
Compute the unique frame of reference.
For each point in scene:
compute its coordinates in the above frame of reference, and look up
any entries in the hash table under these coordinates.
These entries represent points that match the above scene point under
the given transformation. For each entry found:
check if the basis pair in it is consistent:
if so, "cast a vote" (raise a counter) for the pair (model,f.o.r.)
and match the point on the scene to the point in the model.
After finishing going through the basis pairs, cluster the highest - ranking
matches by transformation.
For each cluster:
calculate the transformation (by a least RMSD criterion) given from
all matched basis pairs .
For each model:
Extend the match list (if you took only a part of the scene for the
preceding stages) ,i.e. take the model, map it point by point to the scene
under the calculated transformation, add all consistent matches to the
match list.
give (model, Transformation [,match list]) as output.
The Application of Geometric Hashing to molecular biology.
The method stated above can be applied in the 3D case. This is conceptually
no problem, the only difference is that for determining a frame of reference
uniquely in R3 we need basis triplets, i.e. 3 nonlinear points (one for
the origin, another will determine the unit X axis, and the third will
span a plane ( with orientation) which will give the Y and Z axes.)
So we may take the above scheme, and replace all the basis pairs to
basis triplets.
The difference will, of course, result in higher complexity: The Preprocessing
stage will now have an O(N4 *M) time complexity, While the Recognition
stage will take O(S4) time.
To reduce the complexity , we can :
-
take only triplets that are not 'too small' ( for numeric reasons) or 'too
big' ( these have less chance of appearing in an a molecule with a partial
match to the input).
-
In the case of linear polymers (which is the dominant case in biological
macro-molecules) we can use the linearity , and look only at triplets which
are in the right order in the sequence, or even give some heuristic ( such
as Ci , Ci+2 , Ci+4 , for example, or like taking the Ca
, C , and N atoms from each amino acid.) that will reduce the complexity
to linear ( at the risk of losing some potential solutions).
In the case of Proteins, which is our main concern in this course, we can
significantly reduce complexity of the GH algorithm by using secondary
structure features, such a Alpha Helices or Beta strands. These can be
approximated with straight lines, and 2 non parallel straight lines in
R3 form a basis.
Since these features usually have at least several dozens of Amino Acids,
using them , plus all the "unrelated" Ca atoms can reduce the
input size in 1-2 orders of magnitude, thus greatly cutting both the worst
case and the mean time complexity.
Back to Index.
The Problem of Non rigid bodies, specifically
rotary joints:
Molecules , when they bond with other molecules, tend to change their
shape.
In some cases, these changes can be approximated with rotational movements
around fixed hinges.
This gives us ( C.S. people) the motivation to try and solve the above
problem(s) with an option for movements of parts of the molecule around
fixed hinges.
So, if we have some prior information we can try and give an answer
for the molecule matching problem that will have better correspondence
to the biological (wanted) solution.
Statement of Problem:
Given a database of known molecules, each represented as a (possibly
linearly ordered) set of points in R3 and possible rotary joints
, and an input molecule ( "scene") find the best (set of) choice of:
Molecule in the DB , Rigid transformation and a set of rotations (
on the chosen molecule's joints) ,such that the chosen molecule, under
the transformation and the bending of joints, will match the input molecule
most closely.
We are interested in a Scheme that will "vote" for consistent parts
of a molecule together, not as different pieces, since this would cut down
Post processing, and improve the quality of the solution (we can lower
the bound, which means more possible answers.)
Geometric Hashing with Hinges - the solution:
Let us look in the simple case of one hinge first: in this case, we
have 2 moving parts.
For each (un-occluded) part, there is a rigid Transformation from the
model to the scene, that gives us the part's (invariant) frame of reference.
Also, in this local frame of reference, the translation from the origin
to the axle is invariant.
This gives us the motivation to use local f.o.r.s (basis triplets)
and vote for quartets {model, part, axle, f.o.r.}.
Again , we have 2 stages to the algorithm:
-
Preprocessing stage:
for each part we iterate on all basis triplets of its points.
For each basis triplet we:
Compute the associated frame of reference.
Compute the translation to the Axle (No need for rotation...)
For each point in part we:
Compute its coordinates in the above frame of reference
Use the coordinates as a key for inserting an Entry to a Hash table.
The Entry will include: {
Model name. (if more then one Model)
Part Name.
Frame of Reference
Translation (of f.o.r) to Axle.
Identity of point in model
}
The complexity of this stage is O( N4) where N is the number
of points in each part.
Again, this stage requires no knowledge of the "Scene" and is done
off line.
-
Recognition stage:
Given a scene:
For each ordered triplet of points (in a "small enough" region):
Compute the unique frame of reference.
For each point in scene:
Compute its coordinates in the above frame of reference, and look up
any entries in the hash table under these coordinates.
For each entry found:
check if the basis triplet and the Axle in it are consistent with the
scene: if so , "cast a vote" (raise a counter) for the quartet
(model, part, axle, f.o.r.) and match the point in the scene to the point
in the model.
After finishing going through the triplets, cluster the highest - ranking
matches by transformation and Axle.
For each cluster ,calculate the transformation (by a least RMSD
criterion)
given from all matched triplets.
For a specific part, Extend the match list (if you took only a part
of the scene for the preceding stages).
give (model, part, Transformation [match list]) as output.
As before, Extending the match list simply means this:
For each part of the recognized model:
take each point (not already matched) and transform its coordinates,
then check in the input for a point with "close enough" coordinates: when
(an unmatched) one is found, match it to the one in the model.
The complexity of this stage is O(S4) where S is the number
of points in the scene.
The generalization to the case of a linear chain of parts is obvious, but
the generalization to the case where we have several hinges on the same
part or several parts on the same Axle (some general "graph") give rise
to questions of weighting, i.e. the same triplet should vote to several
hinges, should its vote be the same as a triplet voting for only 2 Axles?
for 1 ?
This is done for a single model, but can just as easily be done for
M models in a DB, where the complexity of both the preprocessing and the
recognition stages (assuming a good hash table) will be linear in
M.
Also ,the above method will work in the special case of no bending
of joints, so that "limited over jointing" will not stop from identifying
the wanted model.The last remark is cautious, since fixing a joint in a
molecule means that a lot of the ("non local") basis triplets will not
be entered to the hash table and therefore will not influence the identification,
so that "greatly over jointing", will cause the algorithm to function poorly.
Back to Index.