Once the three-dimensional structure of a protein has been determined experimentally or predicted in-silico, a central and challenging task of computational structural biology is to identify sites of functional importance such as binding sites of small molecules, proteins or antibodies or sites of post-translational modifications. We introduced for this purpose ScanNet (Spatio-Chemical Arrangement of Neighbors Network) is an interpretable, multi-scale geometric deep learning architecture tailored for protein structures. ScanNet builds representations of atoms and amino acids based on the spatio-chemical arrangement of their neighbors. We trained ScanNet for detecting protein-protein, protein-antibody and protein - intrinsically disordered proteins binding sites and established state-of-the-art performance. The network learns structural motifs of interest directly from raw data; we found that it detects simple, generic structural motifs such as hydrogen bonds or solvent exposed hydrophobic side-chains, whereas others recognize complex, task-specific motifs such as hotspot "O-rings" or transmembrane helixes.

Examples of Protein-protein binding site predictions for homodimers.

Examples of Protein-protein binding site predictions for heterodimers.
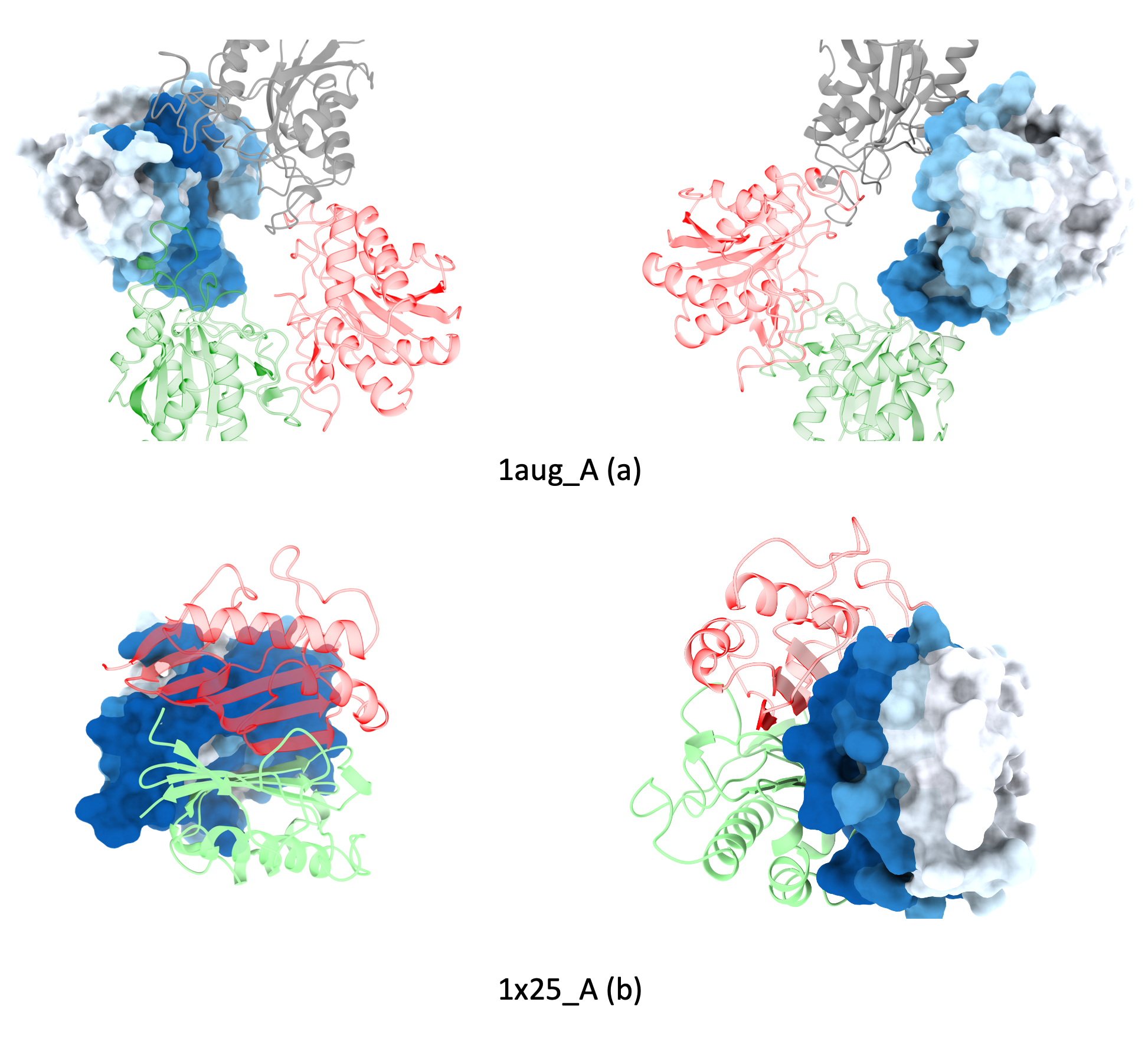
Examples of Protein-protein binding site predictions for homomultimers.
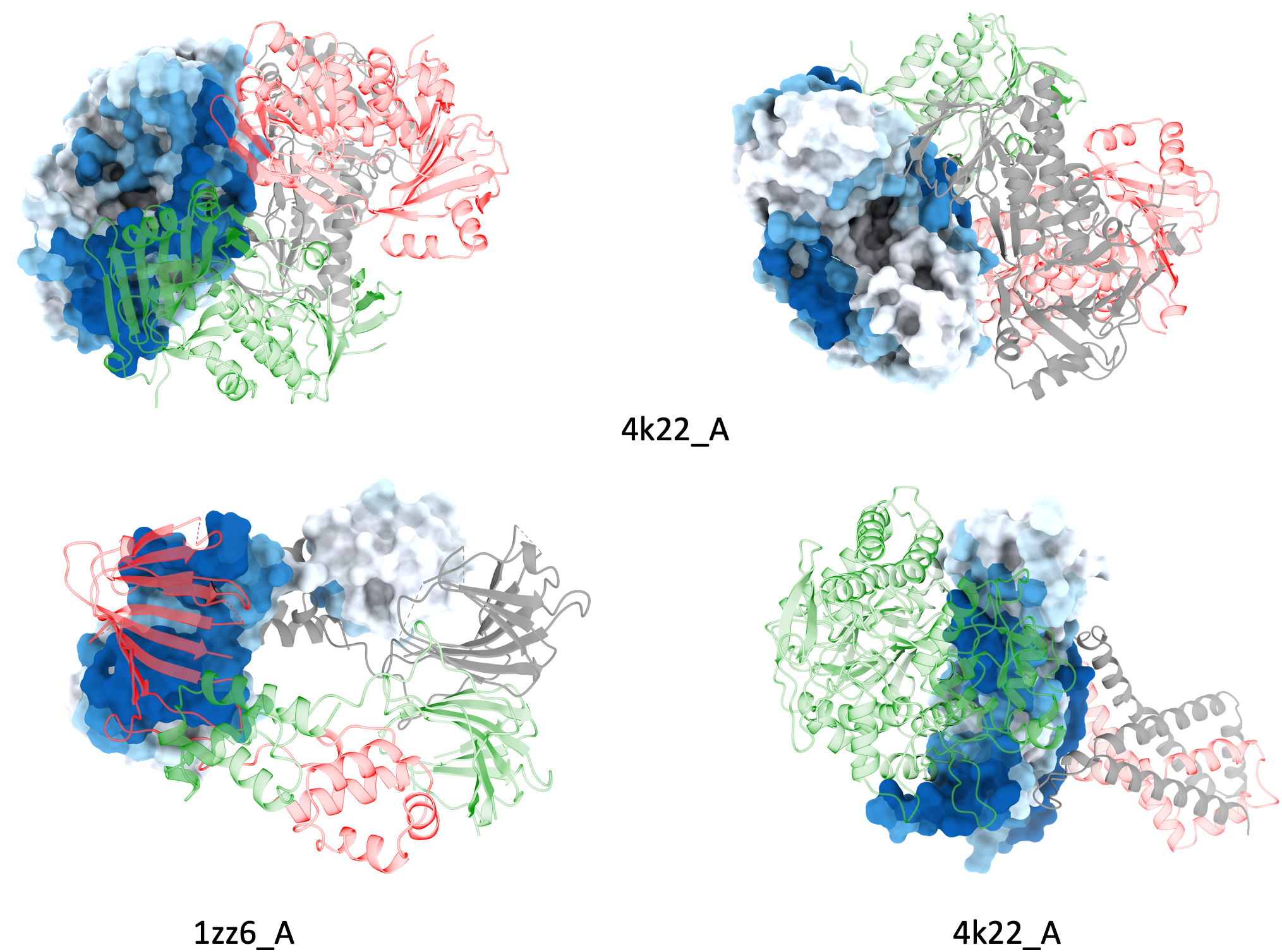
Examples of Protein-protein binding site predictions for heteromultimers.
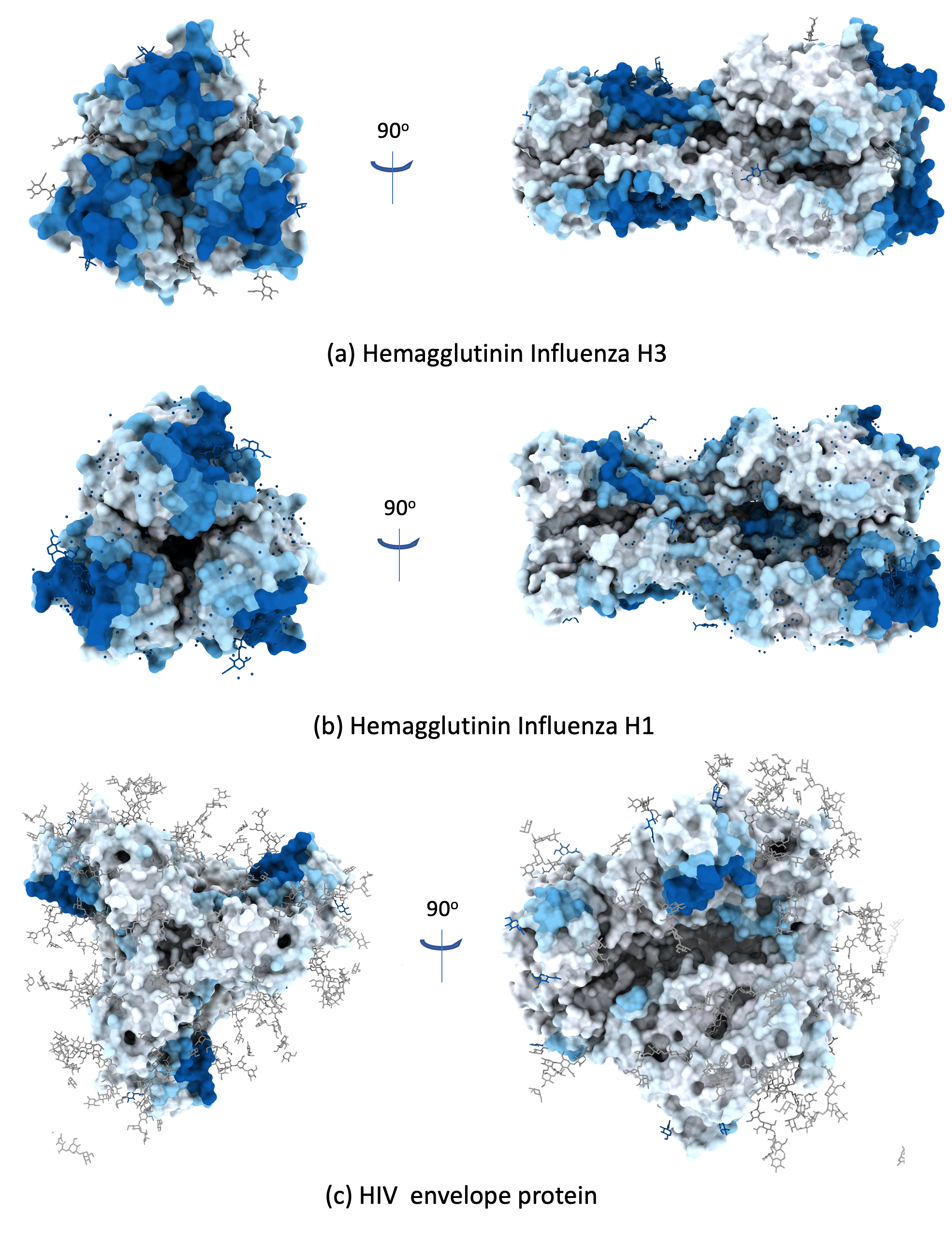
Examples of Protein-antibody binding site predictions for viral envelope proteins.