
Modelling of multimolecular protein complexes
DockStar is an algorithm for modeling of multimolecular protein complexes.
It integrates both high resolution data of the individual subunits and low resolution data, such as the complex interaction graph and chemical cross-links.
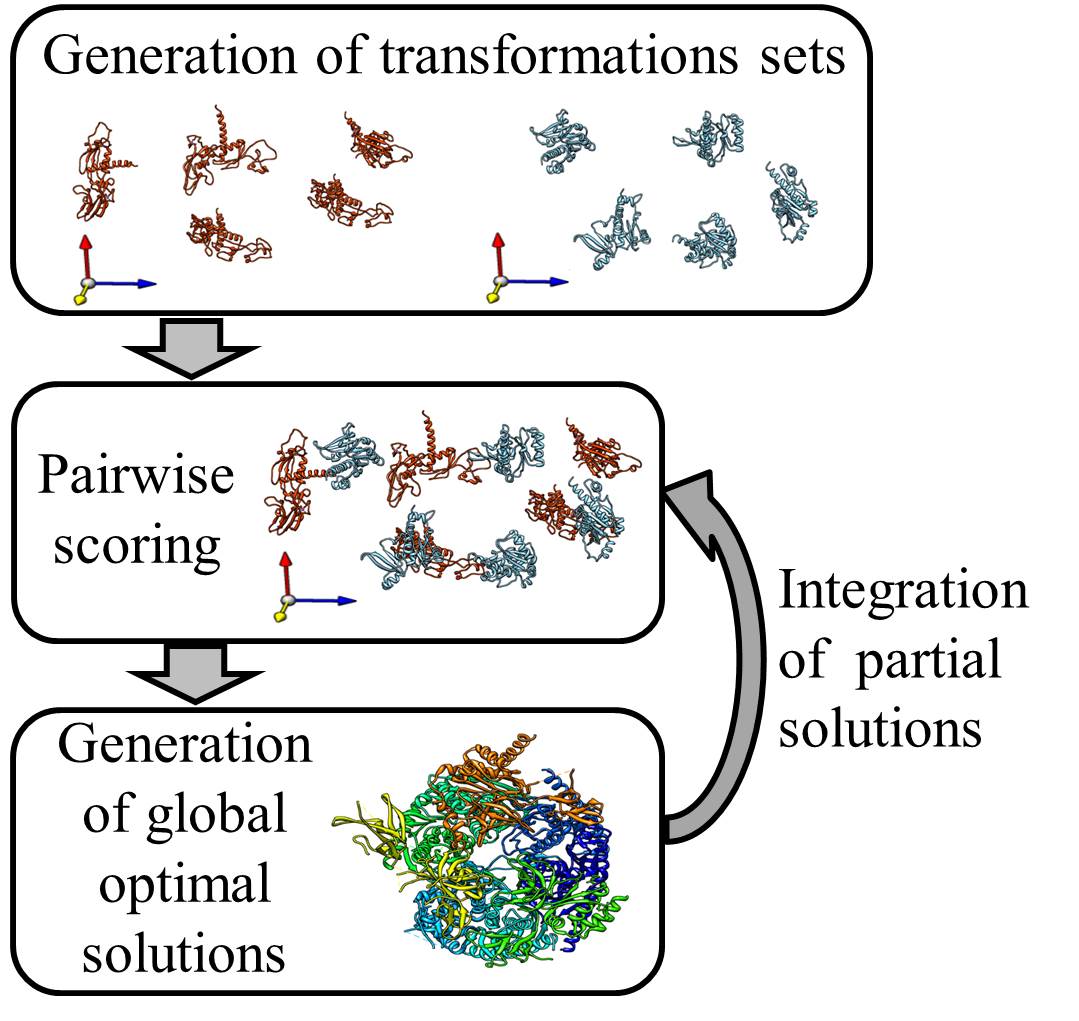
- Retrieving a rigid transformation set for each subunit:
for each subunit a set of alternative transformations is necessary. Each transformation represents an optional location of the subunit in the 3D space. These transformation sets can be generated by several methods, depending on the existing experimental data. If the interaction graph of the complex is available, a set of transformation hypotheses per subunit can be generated by detecting an anchor subunit, which has a high degree in the interaction graph (e.g. many contacting neighbors), and docking its neighbors to this anchor subunit. If a homologous complex is available, these transformations can be generated by homology modeling. Moreover, if a high enough resolution cryo-EM map of the multimolecular assembly is available, one can compute the transformations by fitting to such a map - Pairwise scoring:
each pair of transformations for each two different subunits is scored acccording to its compatibility with the cross links and according to a knowledge based potential. Cross links provide an upper bound on the distance between two residues on the surface of different subunits. A cross link is considered compatible with a transformation pair if the Euclidean distance between the Cα of the linked residues, after executing the transformations, is lower than 30Å. - Choice of a globally optimal solution:
a globally optimal solution is detected for the entire assembly. This solution includes one rigid transformation for each subunit. The transformations are chosen to optimize local interaction score between neighboring subunits and their satisfaction of the experimental cross links constraints. The optimization problem is solved by formulating the task as an Integer Linear Program. The algorithm is tuned to produce a ranked set of (user predefined) K best solutions. - Integration of partial solutions:
In some cases, the structural solution of a protein complex can be calculated iteratively. First, the complex is devided to sub-complexes. Each is solved seperately by DockStar. Top solutions of sub-complexes that share a common subunit can be merged and translated to new transformation sets. These transformation sets will serve as input for a new DockStar run that will generate structural solutions of the whole complex or of a bigger sub-complex.
DockStar webserver implements steps 2 and 3. Hence, the transformation set for each subunit needs to be generated indepndently.
An explicit explanation on how to generate the transformation sets and how to invoke the webserver can be found in the help.
If needed, translation of top solutions of sub-complexes to transformation sets is supported under merge solutions.