| MolAxis is a tool for the identification of high clearance pathways or corridors which represent molecular channels in the complement space of proteins. It is extremely efficient because it samples the medial axis of the complement of the molecule, reducing the problem dimension to two, since the medial axis is composed of surface patches. It is designed to analyze proteins channels, calculate pore dimensions and analyze atom accessibility. MolAxis reads files in the standard Protein Data Bank format (PDB) containing a single frame or multiple frames generated by molecular dynamics (MD) simulations. |
| MolAxis handles two distinct scenarios: It computes channels that connect a single point (like an inner chamber) to the bulk solvent, and it also computes transmembrane (TM) channels. MolAxis has a friendly web interface (see the Web Server tab). It also has a stand-alone version, statically compiled for linux, which can be downloaded from the Download tab. |
| MolAxis Algorithm |
|
We give here a high level description of the algorithm. The algorithm employs computational geometry techniques. Full details of the theory and the algorithm are given in the thesis cited in the reference section below. The algorithm is composed of the following steps:
|
|
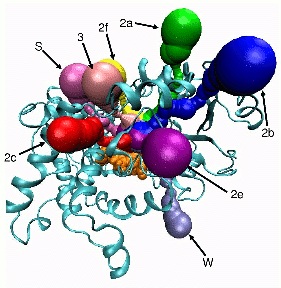
| MolAxis Features |
A short list of the tool features include:
- Finding channels that connect a chamber to the bulk solvent.
- Finding transmembrane channels.
- Graphical display of channels, using Jmol or VMD.
- Channel properties, such as dimension and lining residues.
- Supports a web interface and a stand-alone native linux version.
- Highly accurate and efficient. Robust due to the use of the CGAL library.
| References |
The authors request that all published work which utilizes MolAxis include at least the citation of the paper "MolAxis: A server for Identification of Channels in Macromolecule", as given below.
-
Description of the MolAxis algorithms:
Eitan Yaffe, Dan Fishelovitch, Haim J. Wolfson, Dan Halperin, Ruth Nussinov. "MolAxis: Efficient and Accurate Identification of Channels in Macromolecules." PROTEINS: Structure, Function, and Bioinformatics. Volume 73, Issue 1, October 2008, Pages: 72-86 [link]
-
Description of the MolAxis webserver:
Eitan Yaffe, Dan Fishelovitch, Haim J. Wolfson, Dan Halperin, Ruth Nussinov. "MolAxis: A server for Identification of Channels in Macromolecule". Nucleic Acids Research. Volume 36 (Web Server issue), 1 July 2008, Pages: W210-W215. [link].
-
Analysis of computational geometry properties of the MolAxis algorithm:
Eitan Yaffe, Dan Halperin. "Approximating the Pathway Axis and the Persistence Diagram of a Collection of Balls in 3-Space". Symposium on Computational Geometry (SoCG '08). [pdf] [slides]
-
M.Sc. thesis of Eitan Yaffe:
Eitan Yaffe. "Efficient Construction of Pathways in the Complement of the Union of Balls in R3". MSc. Thesis, Tel-Aviv University, September 2007. [pdf] [slides]