 |
|
| Functional Sites Structural Search
Engine |
[ About SiteEngine][ Server Help][ Download] |
| Recognizes regions on the
surface of one protein that resemble a specific binding site of another. |
Below is a detailed description of the main forms used in the
SiteEngine server.
To speed up the search please click on the stage on interest.
Forms and stages of the SiteEngine
server:
Stage1 - Input molecules definition
Stage2 - Selection of the chain and the binding site
of interest
Stage3 - Process of SiteEngine
Stage4 - Output of SiteEngine
Stage1 - Input
molecules definition:
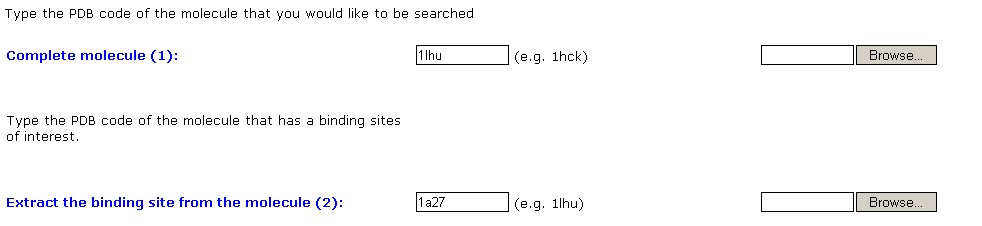
The first stage in activating the SiteEngine is the definition of the
molecules of interest. The definition is performed through the form
below.
The first field specifies the molecules that will be searched for the
binding site of interest. The second field specifies the molecule from
which the binding site will be extracted. If the molecules are
available in the Protein Data Bank (PDB) the PDB codes are to be
specified, otherwise the molecules of interest can be uploaded to our
server. Using the PDB codes speeds up the process, since no file
transfer is required.
Stage2 -
Selection of the chain and the binding site of interest:
First, the user can specify the specific chains of interest that
are to be searched in the complete molecule. This restriction will
significantly speed up the search process.
Second the user must specify the ligand that is present in the binding
site of interest. The region of radius 4.0A around the ligand
will be extracted and used as the searched pattern. SiteEngine will
recognize regions, similar to this pattern, on the surface of the
complete molecule.
Below is an example of a molecule (pdb:1a27) that has two ligands EST
and NAP. By selecting the ligand of interest the user specifies the
regions of interest that his is interested to search in the other
molecule

Stage3 - Process of SiteEngine:
This window shows the process of activation of SiteEngine. The
five main stages are presented and those that are complete are checked
in the checkbox.
In most of the cases the most time consuming stages are the
construction of the surfaces.

Stage4 - Output of SiteEngine:
This window presents the output of the SiteEngine algorithm. The
10 top ranking solutions are presented. These represent the 10 regions
of the surface of the complete molecule that are recognized to be most
similar to the binding site of interest. The file aligned.pdb is the
superimposition of the two molecules by the transformation recognized
by SiteEngine. It can be either downloaded of viewed directly from the
browser. The file contains the superimposition of the two molecules as
well as the functional groups that are recognized to be shared by the
regions. These are also detailed in the output table. To view these
features in the rasmol view please use the following rasmol script.
The output table of SiteEngine presents the details of the common
functional groups.
Below is the description of the columns of the table (click on the
column of interest to jump to a description):
Chain.ID
AminoAcid
Property
Source
Dist
Conserved AA
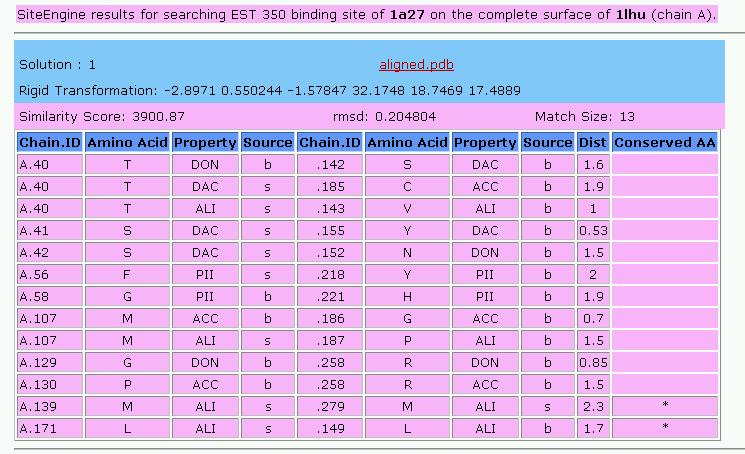
Chain.ID:
The protein chain, followed by the identity of the amino acid
AminoAcid:
The one letter amino acid code. However it must be noted that the
method is based on the physico-chemical properties and does not
consider the identity of the amino acids. These are only displayed for
the convenience of analysis.
Property:
The physico-chemical property that is matched by the algorithm. The
method is based on a representation of each amino acid of a protein as
a set of features that are important for its interaction with other
molecules. The abbreviations of these features are:
DON - Hydrogen bond donor
ACC - Hydrogen bond acceptor
DAC - Hydrogen bond donor and
acceptor (e.g in histidine)
ALI - Aliphatic Hydrophobic
property
PII - Aromatic property (pi
contacts)
Source:
This field specifies whether the matched property is contributed by the
backbone or the side-chain of the amino acid.
The abbreviations are:
b - feature contributed by the
backbone
s - feature contributed by the
backbone
Dist:
The distance in space measured between the matched
features.
Conserved
AA:
Marks the features shared by the two molecules that are contributed by
residues with the same identity of the amino acid.
Reference:
Shulman-Peleg A, Nussinov R, Wolfson HJ, Recognition of functional
sites in protein structures.
J Mol Biol. 2004 Jun 4;339(3):607-33.
Contact: shulmana@tau.ac.il