[About MultiBind]
[Server Help]
[Download]
Recognizes Spatial Chemical Binding Patterns Common to a Set of Protein Structures
| |
|
| [Home] [About MultiBind] [Server Help] [Download] | |
| Multiple Alignment of Protein Binding Sites Recognizes Spatial Chemical Binding Patterns Common to a Set of Protein Structures |
|
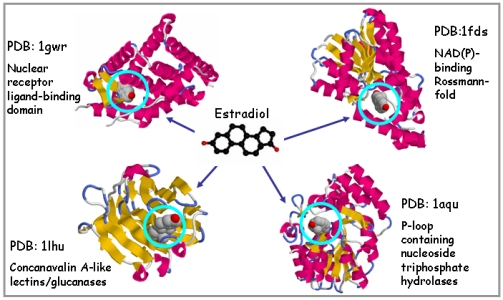
Abstract: MultiBind is a novel computational method, for recognition of binding patterns common to a set of protein structures. It performs a multiple alignment between protein binding sites in the absence of overall sequence, fold or binding partner similarity. MultiBind recognizes common spatial arrangements of physico-chemical properties in the binding sites. These should be important for recognition of function, prediction of binding and drug design. The MultiBind method, applies an efficient Geometric Hashing technique to detect a potential set of multiple alignments of the given binding sites. To overcome the exponential number of possible multiple combinations it applies a very efficient filtering procedure which is heavily based on the selected scoring function. Our method guarantees detection of an approximate solution in terms of pattern proximity as well as cardinality of multiple alignment.
Biological
Motivation:
Question: |