| [Home][Help][About RsiteDB][Download files] |
| Input: The user should specify a PDB code of the protein-RNA complex of interest. |
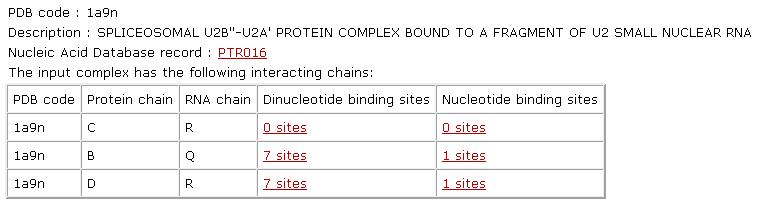
General analysis: Given the input PDB code, RsiteDB will present its description and will detail its interacting protein-RNA chains. For each pair of interacting chains, it will list the number of protein binding sites that accommodate extruded nucleotides and dinucleotides. A nucleotide binding site is defined by the protein Connolly solvent-accessible surface area within 2A from the surface of the RNA nucleotide ring. Only nucleotides with a protein binding site area larger than 3A are considered as protein interacting. Each pair of extruded consecutive nucleotides that interact with the protein, will be considered as dinucleotides and their binding site will be analyzed as a single unit. Binding sites of single nucleotides that are not a part of a pair of consecutive interacting nucleotides are considered as single-nucleotide binding sites. |
 |
Dinucleotide binding sites:
|
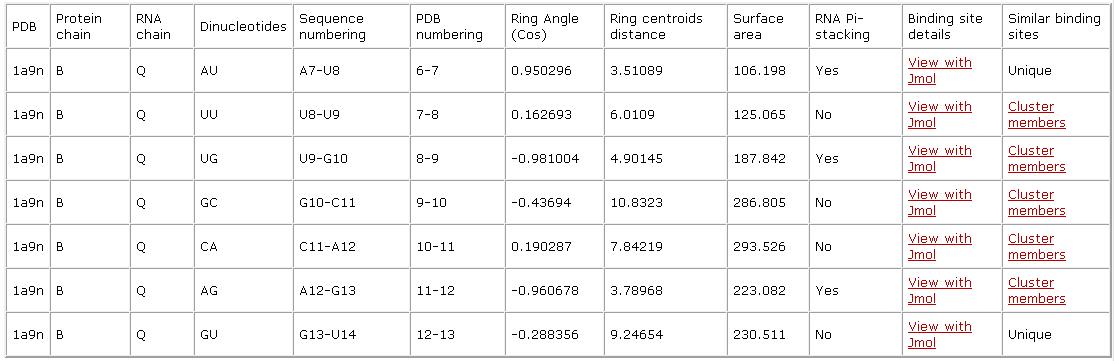 |
| Single nucleotide binding site: Given a nucleotide binding site, that does not have a protein interacting RNA strand neighbor, we analyze it as a single nucleotide binding site. Similarly to dinucleotide binding sites, we detail the nucleotide identity and sequence numbering, where present the numbers listed in the PDB file (PDB numbering) as well as enumerate the nucleotides according their location on the specified RNA chain (Sequence numbering). We detail the binding site surface area and provide a link to a Jmol visualization and a list of binding sites with similar patterns (Cluster members). |
 |
| Cluster members: RsiteDB is based on a classification of the nucleotide and dinuclaotide binding sites. For each binding site we detail its cluster members, which are binding sites with similar spatial physico-chemical binding patterns. Moreover, we present the details of this pattern shared by all cluster members. Specifically, each physico-chemical property is defined by the following description: Amino Acid - The one letter amino acid code. However it must be noted that the method is based on the physico-chemical properties and does not consider the identity of the amino acids. These are only displayed for the convenience of analysis. Type - The type of physico-chemical property that is matched by the algorithm. The method is based on a representation of each amino acid of a protein as a set of features that are important for its interaction with other molecules. The abbreviations of these features are: DON - Hydrogen bond donor; ACC - Hydrogen bond acceptor; DAC - Hydrogen bond donor and acceptor (e.g in histidine); ALI - Aliphatic Hydrophobic property; PII - Aromatic property (pi contacts). Source This field specifies whether the matched property is contributed by the backbone or the side-chain of the amino acid. The abbreviations are: b - feature contributed by the protein backbone atoms; s - feature contributed by the protein side-chain atoms. Same AA: Marks the features shared by the two molecules that are contributed by residues with the same identity of the amino acid. Each solution can be visualized with Jmol. The default view presents the superimposition of all the complexes, with their corresponding nucleotides/dinucleotides and the matched physico-chemical properties. The molecules are colored by model (according to the order of input molecules, the first is cyan, the second is magenta, the third is yellow). The physico-chemical properties (pseudocenters) are represented as balls. Hydrogen bond donors are blue, acceptors - red, donors/acceptors - green, hydrophobic aliphatic - orange and aromatic - gray. The surface points that have a similar location in all the molecules are represented as smaller dots with the same coloring. |