| [Home][Help][About RsiteDB][Download files] |
| Input : The user can specify a PDB code of interest or upload the structure of a target protein. The protein structure can be unbound. If the structure is uploaded it should be in a PDB file format. | |
Chain specification: The input structure is analyzed and the user is prompted to select the protein chain of interested. If all protein chains should be considered the user should select the "All chains" option, but this will slow down the computation. | |
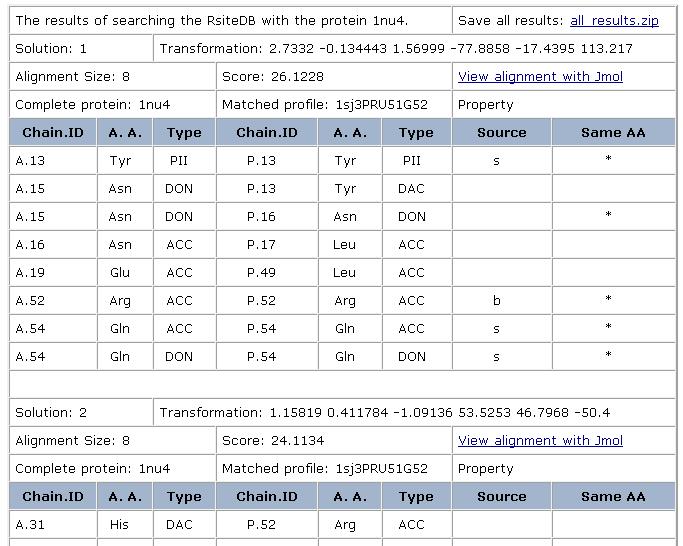
Prediction of dinucleotide binding sites: Here, we
use the created clusters to predict RNA binding sites that accommodate
unpaired extruded dinucleotides. Specifically, given a target protein
structure not used for the classification, we search its surface for
regions similar to the created 3D consensus binding patterns. These
regions are predicted to serve as dinucleotide binding sites. Using
leave-one-out tests, the success rate of these predictions was
estimated to be about 80%. It must be noted that currently we do not
aim to predict whether a protein can bind RNA; rather, given an
unbound RNA binding protein, our goal is to predict its binding sites
and their modes of interaction. In addition, due to a low number of
single nucleotide clusters, currently, we do not use them for the
prediction. Each solution can be visualized with Jmol. The default view
presents the superimposition of all the complexes, with their
corresponding nucleotides/dinucleotides and the matched
physico-chemical properties. The molecules are colored by model
(according to the order of input molecules, the first is cyan, the
second is magenta, the third is yellow). The physico-chemical
properties (pseudocenters) are represented as balls. Hydrogen bond
donors are blue, acceptors - red, donors/acceptors - green,
hydrophobic aliphatic - orange and aromatic - gray. The surface points
that have a similar location in all the molecules are represented as
smaller dots with the same coloring.
|