
Flexible Induced-fit Backbone Refinement in Molecular Docking
About FiberDock
FiberDock is a new method for flexible refinement of docking solution candidates (ref. 1,2). The method models both side-chain and backbone flexibility and performs rigid body optimization on the ligand orientation. The refinement algorithm mimics an induced fit process. The backbone and side-chain movements are modeled according to the binding van der Waals forces between the receptor and ligand. The method uses both low and high frequency normal modes and therefore is able to model both global and local conformational changes, such as opening of binding sites and loop movement. After refining all the docking solution candidates, the refined models are re-scored according to an energy function.
In FiberDock web-server the user can upload/specify PDB codes of two PDB files (receptor and ligand) and provide a list of transformations. Each transformation, when applied on the ligand, produces a candidate docking solution. The candidate solutions for FiberDock can be generated by rigid-body docking methods, such as PatchDock (ref. 4,5), FFT-based methods such as ZDOCK , GRAMM-X , Hex, ClusPro etc. The output is a ranked list of all the refined input solutions. The user can view the complexes in the Jmol applet window and/or download the structures.
Each candidate is subsequently refined by restricted interface side-chain rearrangement, by backbone minimization in the directions of calculated normal modes and by soft rigid-body optimization. The side-chain flexibility is modeled by rotamers and the obtained combinatorial optimization problem is solved by integer linear programming (ref. 6). Following rearrangement of the side-chains, an NMA based backbone refinement procedure is applied. Finally, the relative position of the docking partners is refined by Monte Carlo minimization of the binding score function. The refined candidates are ranked by the binding score. This score includes Atomic Contact Energy (ref. 7), softened van der Waals interactions, partial electrostatics and additional estimations of the binding free energy. The energy function and the procedures of side-chain refinement rigid-body optimization were adopted from the FireDock method (ref. 3).
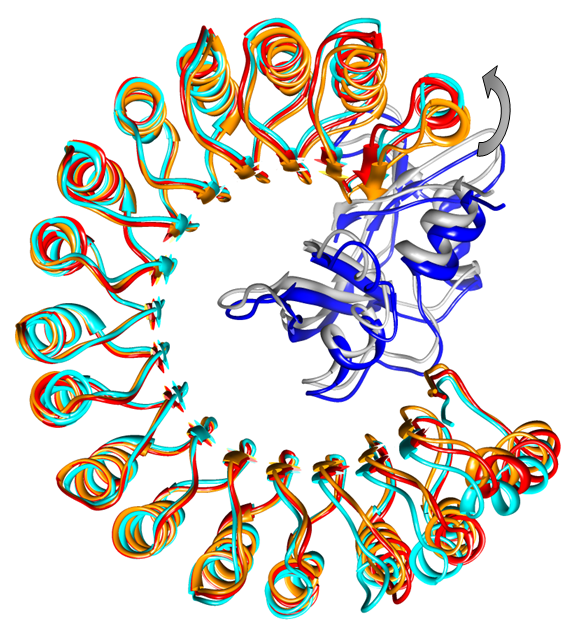
The method (and server) has been extensively and successfully tested on protein-protein docking data-set of ~20 complexes in which the conformation of the receptor's backbone changes upon interaction with the ligand. The image below shows a result of FiberDock refinement of a solution candidate from PatchDock. The unbound structure of the receptors is colored in orange, the bound structure of the receptor is in cyan and the predicted structure of the receptor is in red. The ligand in its native orientation is presented in blue, and the predicted position of the ligand is colored gray. The refinement modeled correctly the backbone movement which occurs upon binding (marked by a gray arrow).
References:
- E. Mashiach, R. Nussinov and H. J. Wolfson. FiberDock: Flexible induced-fit backbone refinement in molecular docking. Proteins 2009 Dec 9;78(6):1503-1519.
- E. Mashiach, R. Nussinov and H. J. Wolfson. FiberDock: a web server for flexible induced-fit backbone refinement in molecular docking. Nucleic Acids Res. 2010 Jul 1;38 Suppl:W457-61.
- N. Andrusier, R. Nussinov and H. J. Wolfson. FireDock: Fast Interaction Refinement in Molecular Docking. Proteins 2007, 69(1):139-59.
- D. Duhovny, R. Nussinov, and H. J. Wolfson. Efficient unbound docking of rigid molecules. In R. Guigo and D. Gusfield, editors, Workshop on Algorithms in Bioinformatics, volume 2452, pages 185-200. Springer Verlag, 2002.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov, H. J. Wolfson. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucl. Acids. Res. 33: W363-367, 2005.
- C. L. Kingsford, B. Chazelle, and M. Singh. Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics, 21(7):1028-1036, 2005.
- C. Zhang, G. Vasmatzis, J. L. Cornette, and C. DeLisi. Determination of atomic desolvation energies from the structures of crystallized proteins. J Mol Biol, 267(3):707-726, April 1997.